Sickle cell disease is caused by a complex and often misunderstood condition that affects millions around the globe. For patients, caregivers, and healthcare professionals, grasping the intricate web of factors that contribute to this disease is essential for comprehensive care and effective management. In this in-depth discussion, we will explore the underlying causes of sickle cell disease is caused by, providing insights that illuminate the path toward better understanding and treatment.
A Genetic Tapestry: The Roots of Sickle Cell Disease
At the core of sickle cell disease is caused by lies a genetic mutation within the hemoglobin gene, specifically in the beta-globin chain. This inheritance pattern means that the disease is passed down from parents to children. Understanding this genetic basis is crucial for families navigating a diagnosis and for researchers seeking to untangle the disease’s complexities.
Unraveling the Genetic Mutation
A single nucleotide change in the DNA sequence results in the production of abnormal hemoglobin known as hemoglobin S. This simple substitution is responsible for the altered shape and function of red blood cells. The presence of two abnormal beta-globin genes, one from each parent, leads to the most severe form of sickle cell disease while having only one can cause sickle cell trait, a milder condition.
Prevalence and Distribution
Sickle cell disease is caused by is most commonly found in populations with sub-Saharan African, South American, Central American, and some Mediterranean heritages. The disease’s geographical distribution mirrors the historical and contemporary movement of these populations. This point of origin explains why the condition is frequently referred to as “African” or “Black” sickle cell anemia, despite its global presence.
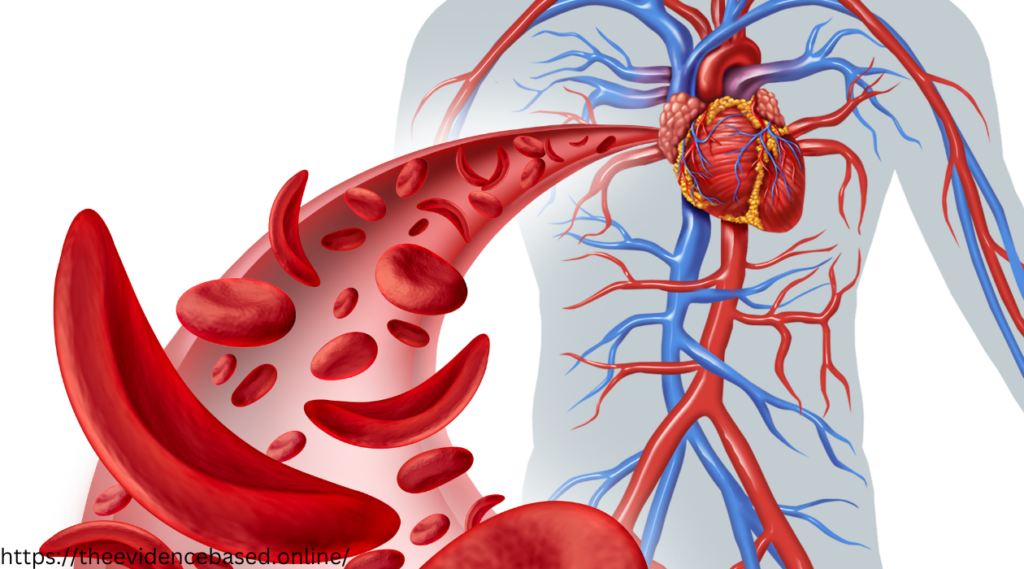
The Molecular Culprits: Hemoglobin S and Its Consequences
The normal function of hemoglobin is to transport oxygen from the lungs to the rest of the body. In sickle cell disease, the presence of hemoglobin S leads to a cascade of events that culminate in the characteristic sickle-shaped red blood cells.
Hemoglobin and Oxygen Transportation
The unique structure of hemoglobin allows it to bind to oxygen. In healthy individuals, this oxygenated hemoglobin maintains the pliable, discoid shape of red blood cells, permitting easy flow through blood vessels. When severe deoxygenation occurs, hemoglobin S triggers red blood cells to stiffen and distort, a reaction known as sickling.
The Consequences of Sickled Cells
The sickled form of red blood cells causes them to become inflexible and adhesive. These altered cells can be inefficient at oxygen delivery, contribute to vascular occlusion, and have a shortened lifespan. The accumulation and adhesion of sickled cells can lead to painful vaso-occlusive crises, acute chest syndrome, and increased susceptibility to infections.
Navigating the Symptoms and Sickle cell disease is caused
The impact of sickle cell disease is far-reaching, affecting various systems and organs in the body. From infancy through adulthood, individuals with sickle cell disease face an array of symptoms and potential complications that require vigilance and management.
Anemia and Chronic Pain
The destruction of red blood cells and their shortened lifespan lead to chronic anemia, a common feature of sickle cell disease. This anemia can manifest as weakness, fatigue, and shortness of breath. The most well-known and managing pain is a complex challenge for both patients and healthcare providers. Vaso-occlusive crises can occur suddenly and are characterized by debilitating pain, often requiring hospitalization.
Organ Damage and Complications
Chronic organ damage is a significant concern for individuals with sickle cell disease. The repeated episodes of vascular occlusion can lead to damage in various organs, including the spleen, kidneys, and lungs. Additionally, the increased risk of stroke, leg ulcers, and priapism adds layers to the management of this multifaceted condition.
The Diagnostic Odyssey and Ongoing Management of Sickle Cell Disease
Early diagnosis and consistent management are critical in mitigating the effects of sickle cell disease. From laboratory tests to ongoing therapies, the journey of a person with sickle cell disease is one that necessitates a partnership between the patient and their healthcare team.
Laboratory Diagnostics
Several tests are employed to diagnose sickle cell disease, including hemoglobin electrophoresis, complete blood count (CBC), and peripheral blood smear examination. These tests identify the presence of sickle hemoglobin and reveal the characteristic changes in red blood cell morphology.
Therapeutic Strategies & Sickle cell disease is caused
The treatment landscape for sickle cell disease is evolving rapidly. Hydroxyurea has been a game-changer in reducing the frequency and severity of vaso-occlusive crises. Other approaches, such as blood transfusions, stem cell transplants, and emerging therapies like gene editing, are offering new hope and possibilities for those with sickle cell disease.
Nurturing a Life with Sickle Cell Disease: Lifestyle Considerations and Self-Care
Living with sickle cell disease requires dedication to a healthy lifestyle, understanding one’s own body, and a supportive network. Patients, caregivers, and healthcare professionals must work together to create sustainable care plans that address the unique needs of each individual with sickle cell disease.
The Role of Diet and Exercise
A balanced diet and regular exercise can play a significant role in managing the symptoms of sickle cell disease. Proper nutrition supports overall health, while appropriate exercise can help maintain physical strength and stamina. Avoiding triggers like extreme temperatures and dehydration is also crucial in preventing crises.
Psychological and Social Support
The psychological impact of living with sickle cell disease should not be underestimated. Patients benefit from mental health support, as the disease can be isolating and the chronic nature of the pain may lead to depression and anxiety. A robust support system, including patient advocacy groups, can provide a sense of community and belonging.
Pioneering the Future: Research and Potential Breakthroughs
The future of sickle cell disease management holds the promise of significant advancements. Ongoing research efforts are focused on several key areas, to improve outcomes and potentially find a cure.
Gene Therapy and Stem Cell Research
Gene therapy and stem cell research offer revolutionary approaches to sickle cell disease. Clinical trials exploring the use of gene editing technologies and stem cell transplants provide glimpses of a future where inherited genetic conditions like sickle cell disease may be corrected at the source.
Holistic Approaches and Quality of Life
In addition to medical interventions, a holistic approach that considers all aspects of a patient’s life is vital. Quality-of-life studies are examining the impact of sickle cell disease on education, employment, and social well-being, intending to address these challenges through multidisciplinary interventions.
Conclusion: The Imperative of Understanding the Causes of Sickle Cell Disease
For individuals affected by sickle cell disease, their families, and the broader healthcare community, understanding the causes and mechanisms of the condition is fundamental. It paves the way for compassionate care, informed decision-making, and a continued commitment to research and innovation.
Challenges abound in the realm of sickle cell disease, but with a collective effort and the resilience of those touched by the condition, we move closer to a world where the impact of sickle cell disease is mitigated and, ultimately, eradicated. This deeper understanding, combined with ongoing advocacy and support, will shape the future of sickle cell care and, one day, the story of its conquest.